A substrate envelope approach to examine drug resistance potential of SARS-CoV-2 Mpro
A team of scientists from the United States has recently defined the substrate envelop of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The study is currently available on the bioRxiv* preprint server while awaiting peer review.
Study: Defining the Substrate Envelope of SARS-CoV-2 Main Protease to Predict and Avoid Drug Resistance. Image Credit: Mark Umbrella/Shutterstock
Background
SARS-CoV-2, the causative pathogen of coronavirus disease 2019 (COVID-19), is an enveloped, positive-sense, single-stranded RNA virus of the human beta-coronavirus family. The genome of SARS-CoV-2 contains multiple polyproteins that are cleaved by viral proteases to release individual proteins. The viral main protease plays a crucial role in cleaving these sites, which is essential for viral replication. Thus, any antiviral drugs that directly prevent viral cleavage are considered potent inhibitors of viral growth. In this context, clinical studies investigating the antiviral efficacy of main protease inhibitors have shown promising results.
Viral proteases are known to bind protein cleavage sites with diverse amino acid sequences through a conserved three-dimensional structure (substrate envelop). Therefore, viral inhibitors that occupy the same space as endogenous substrates within the substrate envelope are less likely to develop resistance due to viral mutations.
In the current study, the scientists have analyzed nine high-resolution cocrystal structures of SARS-CoV-2 main protease with viral cleavage sites to define the structural basis of substrate recognition.
Important observations
The crystal structure analysis of the main protease with nine substrates and six product complexes (product structures bound to cleaved N-terminal side of the substrate) revealed that the natural cleavage sequences are mostly not conserved. Full conservation was noticed only for glutamine at the P1 position and a hydrophobic leucine/phenylalanine/valine at the P2 position.
The viral main proteases were found to crystalize as a homodimer. Of nine complexes, six had a dimer in the asymmetric unit, with both active sites occupied with a substrate. The other three complexes had a monomer in the asymmetric unit.
Further analysis of the cocrystal structures revealed that the substrate peptide is extended along the main protease active site with the scissile bond. The residues near the scissile bond participated in the molecular interactions between substrate and main protease. While the N-terminal side of the substrate showed full conservation in all structures, the C-terminal residues showed diverse binding modes.
The substrate – protease interaction was stabilized through multiple hydrogen bonds that were formed between the substrate peptides and main protease active sites. An extensive network of backbone–backbone hydrogen bonds was observed in the structures, which determined the substrate specificity of the main protease.
Further analysis of substrate – protease pair revealed that the residues on the active site surface of the main protease could tolerate a wide variety of mutations. However, the activity and substrate interactions remained primarily unchanged.
SARS-CoV-2 main protease substrate envelop
Regarding substrate specificity, the main protease was found to recognize a conserved shape of the substrate, despite differences in the sequence. This conserved shape defines the substrate envelop of the main protease. The analysis of the most conserved cleavage site (non-structural protein 8 – 9 substrates) between coronaviruses revealed perfect fitting of the site within the substrate envelop. Some highly conserved regions between all substrate complexes were identified despite differences in amino acid sequences. This high conservation determines the specificity of substrates.
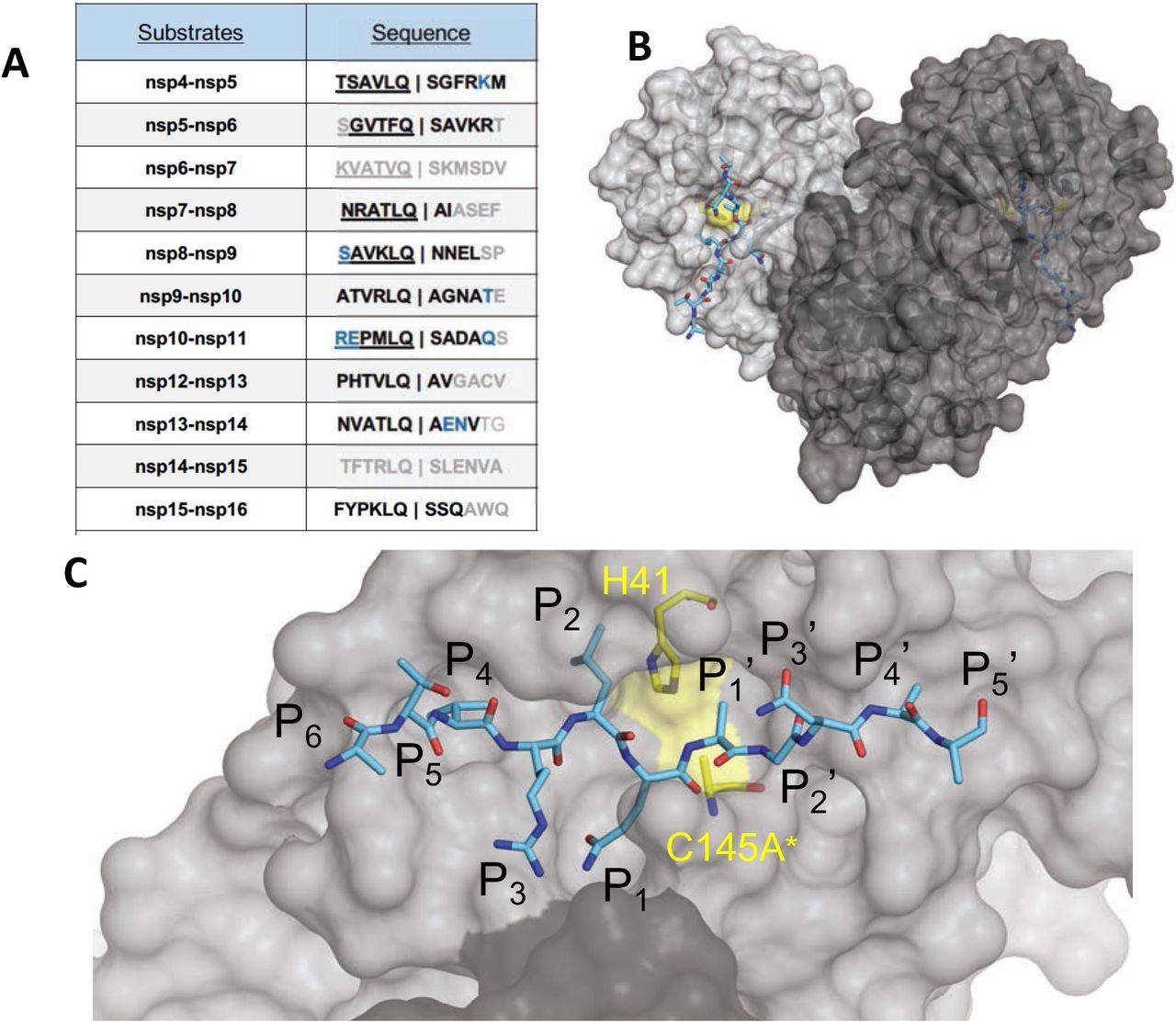
The amino acid sequences and binding of substrates to SARS-CoV-2 Mpro active site. (A) Viral polyprotein cleavage sites processed by Mpro to release non-structural proteins (nsp). The one-letter amino acid codes of cleavage site sequences, where bold letters indicate fully resolved residues and blue are stubbed side chains in the cocrystal structures. Underlined N-terminal sequences correspond to product complexes with independently determined cocrystal structures. (B) Crystal structure of SARS-CoV-2 Mpro with a substrate peptide (nsp9-nsp10) bound at the active site of both monomers (light and darker gray). The peptide is depicted as cyan sticks and the catalytic dyad is colored yellow. (C) Close-up view of one of the active sites in panel B, with the protease in surface representation. The asterisk indicates catalytic cysteine was mutated to prevent substrate cleavage. The cleavage occurs between positions P1 and P1′.
Main protease inhibitors and resistance mutations
The canonical sequence sites (outside the substrate envelop) from where inhibitors protrude to contact main protease resides are highly vulnerable as mutation-induced changes in these sites can potentially reduce inhibitor binding without affecting substrate binding and cleavage.
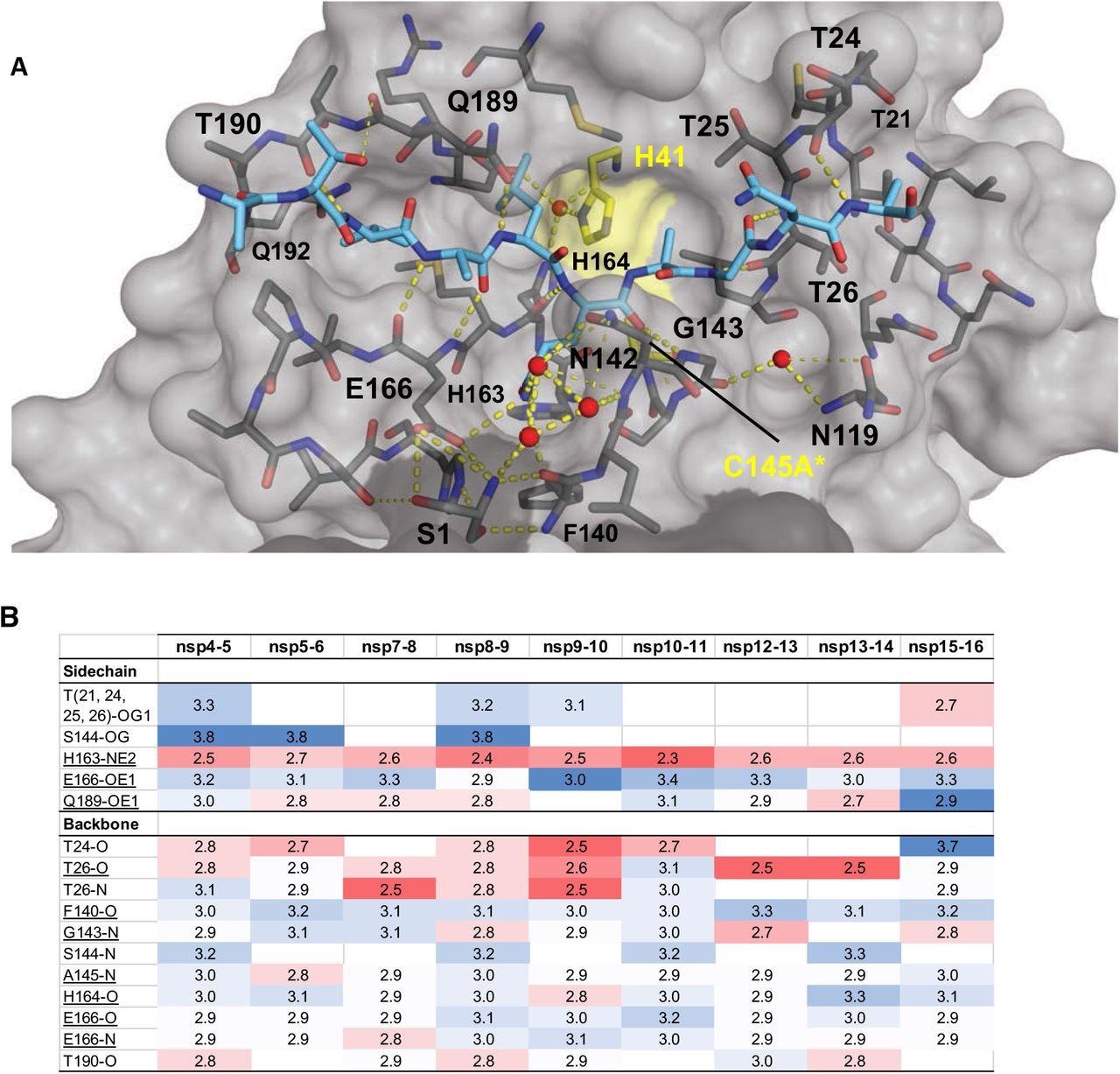
Intermolecular hydrogen bonds in Mpro substrate cocrystal structures. (A) Hydrogen bonds between bound nsp9-nsp10 substrate and Mpro. The substrate peptide is depicted as cyan sticks and the protease is in gray surface representation with the catalytic dyad colored yellow. Yellow dashed lines indicate hydrogen bonds (thicker lines for stronger bonds with distance less than 3.5 Å) and red spheres denote conserved water molecules. Serl depicted as sticks belongs to the other monomer (shown in darker gray). (B) Hydrogen bonds that are conserved in three or more substrate complexes, underlined completely conserved, top interacting with Mpro sidechains and bottom with Mpro backbone atoms, color coded by the closeness of the hydrogen bond.
Four SARS-CoV-2 main protease inhibitors were analyzed in the study, which revealed a diverse pattern of binding with the main protease active site. The three most flexible residues were identified, which showed variations in conformations depending on the inhibitor type. These variations were primarily observed in the substrate envelope sites where the inhibitors protruded. These sites were not conserved among coronaviruses and could serve as potential regions for resistance mutations.
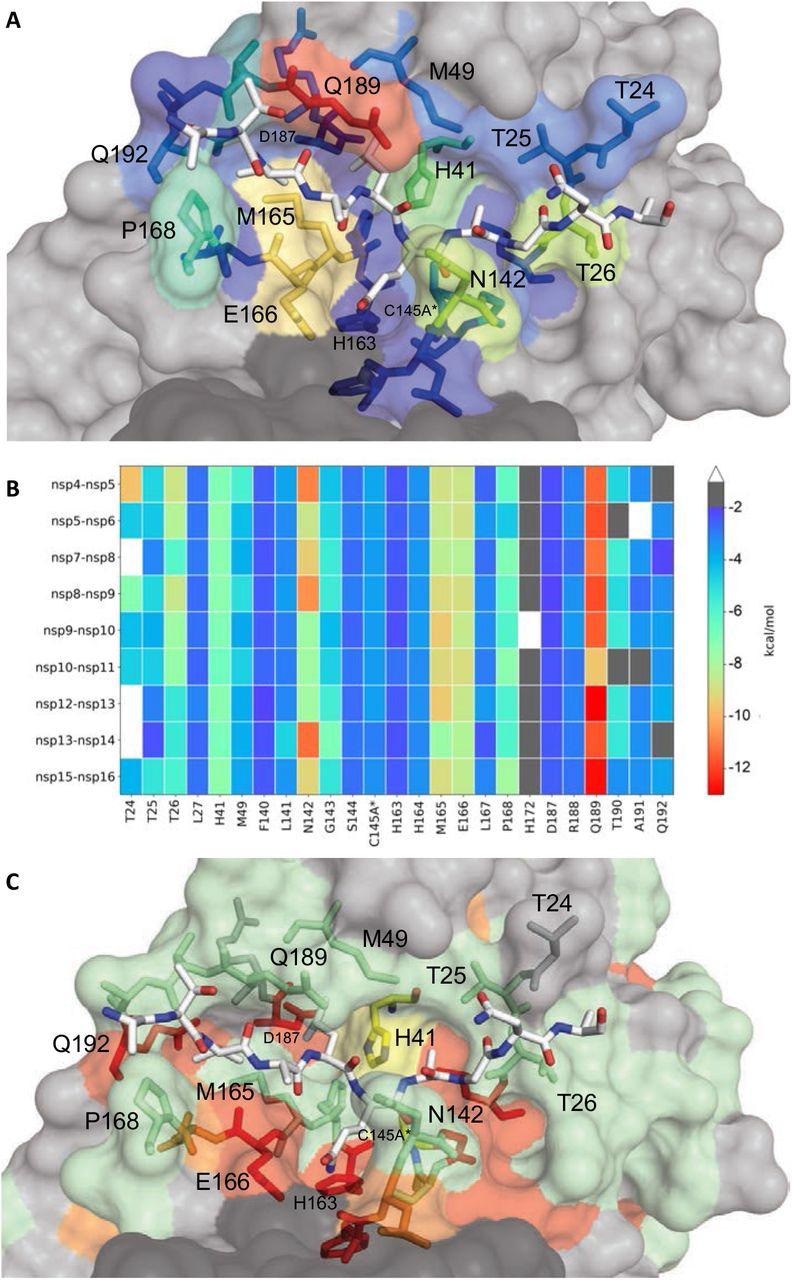
Extent of substrate interactions and conservation of Mpro surface residues. (A) Close-up view of the nsp9-nsp10 substrate bound to Mpro active site in the cocrystal structure where the substrate peptide is depicted as white sticks and the protease is in surface representation. The protease residues are colored according to the extent of van der Waals interactions with the substrate, with warmer colors indicating more interaction. (B) Conservation of substrate-protease van der Waals interactions among the 9 cocrystal structures determined. Heat map coloring by the extent of van der Waals contact by residue. (C) Amino acid sequence conservation of Mpro between 7 (Figure S4) coronaviral species depicted on the structure where surface residues conserved in all 7 (red), 5-6 (orange), 3-4 (green) and less than 3 (highly variable; gray) sequences are indicated by color.
Study significance
The study describes the structural basis of interaction between SARS-CoV-2 main protease and its substrates, i.e., protein cleavage sites. Furthermore, the study reveals that main protease inhibitors that fit within the substrate envelope of main protease are less likely to develop resistance as viral mutations affecting the binding of these inhibitors would also impair the binding between main protease and its substrates.
*Important notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Shaqra AM. 2022. Defining the Substrate Envelope of SARS-CoV-2 Main Protease to Predict and Avoid Drug Resistance. bioRxiv. https://www.biorxiv.org/content/10.1101/2022.01.25.477757v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Coronavirus, Coronavirus Disease COVID-19, covid-19, Cysteine, Drugs, Efficacy, Genome, Glutamine, heat, Leucine, Mutation, Pathogen, Peptides, Phenylalanine, Protein, Respiratory, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Structural Protein, Syndrome, Valine, Virus

Written by
Dr. Sanchari Sinha Dutta
Dr. Sanchari Sinha Dutta is a science communicator who believes in spreading the power of science in every corner of the world. She has a Bachelor of Science (B.Sc.) degree and a Master's of Science (M.Sc.) in biology and human physiology. Following her Master's degree, Sanchari went on to study a Ph.D. in human physiology. She has authored more than 10 original research articles, all of which have been published in world renowned international journals.
Source: Read Full Article


