Overactive enzyme causes hereditary hypertension
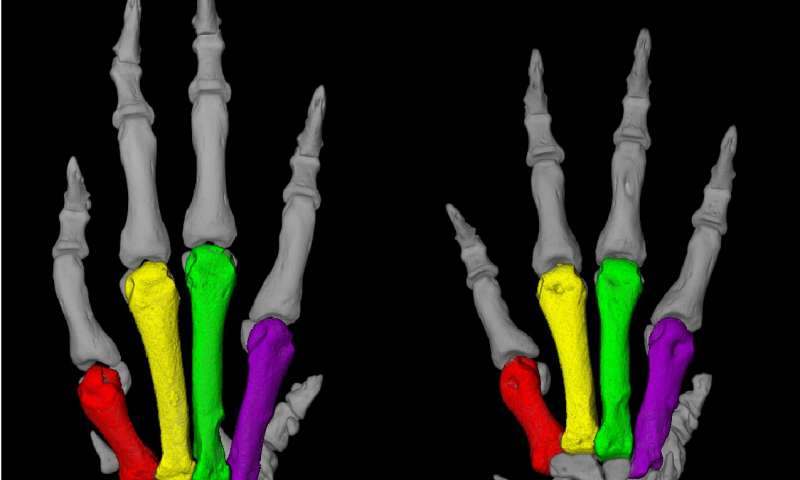
A Turkish family from a village near the Black Sea caught the attention of medical researchers in the early 1970s, when a physician discovered that many members of this large family had both unusually short fingers and astronomically high blood pressure, sometimes twice as high as that of healthy people. Those affected die around the age of 50, usually due to a stroke.
Some 20 years later, a group of researchers at the Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), led by Professor Friedrich Luft and Dr. Sylvia Bähring, began to study this mysterious phenomenon. It proved to be no easy task. Not until May 2015 were the researchers able to report in the journal Nature Genetics that they had found an altered gene in all patients who were affected by the hypertension and brachydactyly (HTNB) syndrome—i.e., high blood pressure and abnormally short digits. The genetic disorder is also known as Bilginturan syndrome, after its Turkish discoverer.
The genetic makeup encodes an enzyme called phosphodiesterase 3A, or PDE3A for short, that regulates both blood pressure and bone growth. The gene mutation that Luft and his team had discovered causes the enzyme to be more active than usual.
Researchers provide the missing evidence
There was no evidence that definitely shows that the mutated PDE3A causes Bilginturan syndrome, which has since been discovered in other families around the world. An international group of 40 researchers from Berlin, Bochum, Limburg, Toronto (Canada) and Auckland (New Zealand) has now supplied this evidence in the journal Circulation. Participating in the study were research groups from the MDC and Charité – Universitätsmedizin Berlin, including teams led by Professors Luft, Michael Bader, Maik Gollasch and Dominik N. Müller as well as Dr. Arndt Heuser and Dr. Sofia Forslund. The last author of the paper is Dr. Enno Klußmann, head of the MDC’s Anchored Signaling Lab.
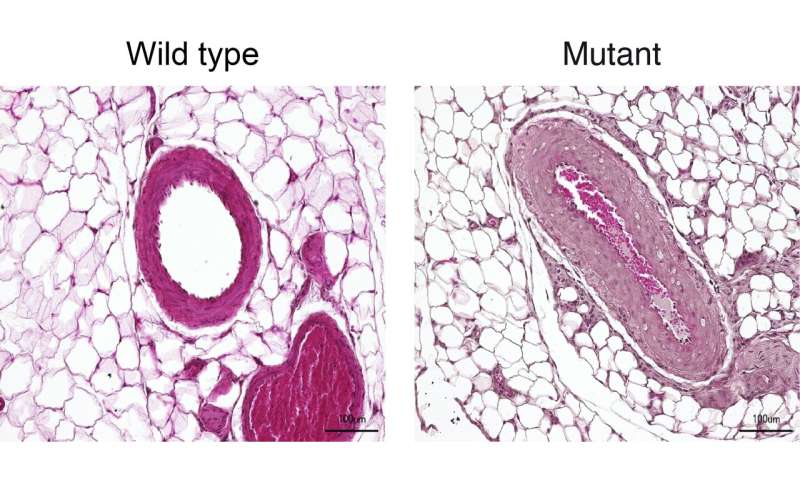
“We mainly worked with two animal models,” reports Dr. Lajos Markó, the paper’s co-lead author along with Maria Ercu. One of the models consisted of genetically modified mice in which the human enzyme PDE3A in the smooth muscle cells of the vessel walls was overactive due to the gene alteration. “These animals exhibited extremely high blood pressure as compared to the control animals,” Markó says.
Genetically modified rats recapitulate the genetic disorder
But what proved more interesting to the scientists was a rat model created by the Bader Lab using CRISPR-Cas9 technology. With the help of the gene-editing tool, the team had altered nine base pairs in a region of the PDE3A gene that is mutated in the syndrome, a so-called mutation hot spot. The resulting enzyme differed from the normal variants with respect to three amino acids. “And just as in the patients, this tiny change increased the activity of the enzyme,” Ercu says.
“The rats resembled human patients to a truly extraordinary degree,” Ercu adds. “They not only suffered from high blood pressure, but the toes on their forefeet were significantly shortened—similar to the fingers of people with the syndrome.” And using micro-computed tomography, the researchers discovered a prominent loop in the brain vessels of the rats that is also found in people with the syndrome. “Our rat model provides, in my view, definitive proof that the syndrome is caused by a mutation in the PDE3A gene,” Klußmann says.
The researchers have even developed an approach for treating this inherited form of high blood pressure. “There is a drug called riociguat that is already approved as a therapeutic for pulmonary hypertension,” Klußmann says. It activates an enzyme that produces a signaling molecule, which in turn dampens down an overactive PDE3A. “The blood pressure of rats to which we administered a derivative of riociguat dropped to a normal level,” Klußmann reports. There are already other PDE3A inhibitors on the market, but they are not suitable for long-term therapy due to their side effects.
Klußmann now wants to take a closer look at how the mutated PDE3A interacts with other protein molecules. Stronger interaction with certain adaptor proteins, he says, could cause cells of the vessel walls to replicate at an increased rate.
Source: Read Full Article

